Research Activities
Disruption of protein trafficking is the molecular basis of many human
hereditary and degenerative diseases. Research in my laboratory is focused on
elucidating the molecular components of these protein sorting and vesicle
trafficking machineries. The endoplasmic reticulum (ER) is the first entry
point to the secretory pathway for secretory and membrane proteins. The ER
exerts a quality control role to ensure that only properly folded proteins are
allowed to progress to the Golgi complex. These properly folded proteins are
sequentially moved from one membrane compartment to another by transport
vesicles. They are loaded as cargoes into transport vesicles along with
regulatory proteins that are required for their delivery to the intended target
organelle. We have previously used a molecular approach to identify a novel
protein which named Prenylated Rab Acceptor (PRA) that regulates an early
transport step. We also identified a previously known protein, called VAMP-associated
protein (VAP), and found that VAP regulates loading of protein cargoes into
transport vesicles. There are two VAP genes in human known as VAPA and VAPB. A
single amino acid substitution in VAPB causes a late-onset, familial form of
Amyotrophic Lateral Sclerosis (commonly referred to as Lou Gehrig's disease).
Mutant VAPB forms insoluble aggregates that cause the ER to form large membrane
structures that trap protein cargoes moving through the secretory pathway. Our
research focuses on the molecular mechanisms by which mutant VAPB induces
formation of these large membrane structures, and ways to mitigate the cellular
effects of these structures.
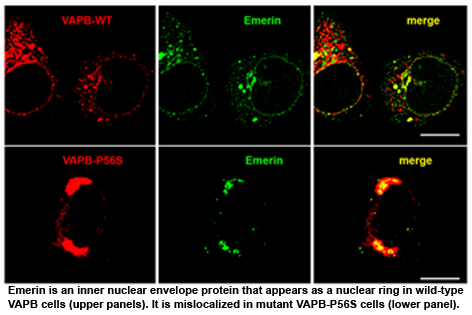
We recently found that expression of mutant VAPB also causes a nuclear
envelope defect due to retention of nuclear envelope and pore proteins in the
abnormal membrane structures generated by mutant VAPB (Fig. 1). VAPB is
required for final delivery of nuclear pore components to the nuclear envelope.
This not only highlights a new mechanism by which proteins reach the nuclear
envelope, but the progressive deterioration of nuclear pores in the absence of
VAPB also provides a mechanism for the age-dependent onset of ALS. Damage or
natural deterioration of pore components is a hallmark of age-dependent human
diseases. This age dependent deterioration is partly due to the very low
turnover of some pore proteins, and synthesis of new pores only occurs shortly
after cell division. Together, this renders non-dividing cells, such as
neurons, more susceptible to age-dependent deterioration of the pores. Loss of
VAPB function further reduces the cell’s ability to make new pores or replace
damaged pores, leading to enhance rate of deterioration. Our current research
seeks to elucidate the molecular mechanism of mutant VAPB-induced disruption of
transport to the nuclear envelope and to design reagents that can restore pore
transport to the nuclear envelope.